| Rabbit Anti-Beta galactosidase antibody |
| 反应物种(预测) |
Chicken,Dog,Pig,Cow,Horse |
| 产品应用(已验证) |
WB |
| 产品应用(可尝试) |
ELISA |
| 推荐稀释比例 |
WB=1:500-1000,Elisa=1:5000-10000, |
| 研究领域 |
细胞生物,免疫学 |
| 标签 |
Array |
-
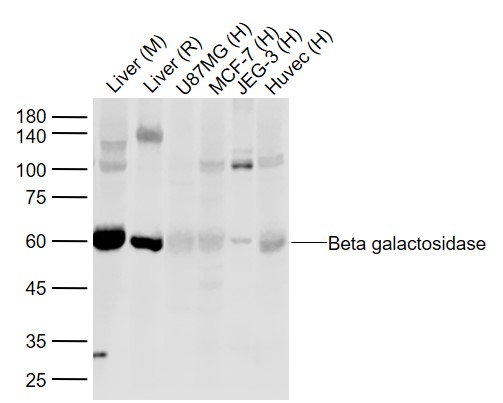
Sample:
Lane 1: Liver (Mouse) Lysate at 40 ug
Lane 2: Liver (Rat) Lysate at 40 ug
Lane 3: U87MG (Human) Cell Lysate at 30 ug
Lane 4: MCF-7 (Human) Cell Lysate at 30 ug
Lane 5: JEG-3 (Human) Cell Lysate at 30 ug
Lane 6: Huvec (Human) Cell Lysate at 30 ug
Primary: Anti-Beta galactosidase (bs-4960R) at 1/1000 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 60 kD
Observed band size: 60 kD
RRID:RRID
产品名称:Rabbit Anti-Beta galactosidase antibody
别名: Acid beta galactosidase;
Acid beta-galactosidase;
Beta galactosidase 1;
Beta-galactosidase;
BGAL_HUMAN;
EBP;
EBP, included;
Elastin receptor 1 (67kD);
Elastin receptor 1 67kDa;
Elastin receptor 1;
Elastin receptor 1, included;
Elastin-binding p
中文名称:β半乳糖苷酶抗体
英文名称:Rabbit Anti-Beta galactosidase antibody
抗体来源: Rabbit
克隆类型:多克隆
细胞定位:细胞浆
性 状:Liquid
亚 型:IgG
纯化方法:affinity purified by Protein A
保存条件:Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles.
免 疫 原:KLH conjugated synthetic peptide derived from human Beta galactosidase
抗原表位:301-380/677
SWISS:P16278
Gene ID :2720
Human Gene ID:2720
Beta galactosidase is coded by a gene (lac z) in the lac operon of Escherichia coli. It is a metalloenzyme that splits lactose into glucose and galactose. It hydrolyzes terminal, non-reducing beta-D-galactose residues in beta-D-galactosides. Activation by cations seems to be substrate dependent. K+, Na+, NH4+, Rb+, Cs+ and Mn++ all activate enzyme activity based upon the substrate used.
Function:Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans.
Isoform 2 has no beta-galactosidase catalytic activity, but plays functional roles in the formation of extracellular elastic fibers (elastogenes
Subcellular Location:Isoform 1: Lysosome. Isoform 2: Cytoplasm, perinuclear region. Note=Localized to the perinuclear area of the cytoplasm but not to lysosomes.
DISEASE:Defects in GLB1 are the cause of GM1-gangliosidosis type 1 (GM1G1) [MIM:230500]; also known as infantile GM1-gangliosidosis. GM1-gangliosidosis is an autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoprotei
Similarity:Belongs to the glycosyl hydrolase 35 family.
Important Note:This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
 400-901-9800
400-901-9800


 说明书
说明书 联系我们
联系我们 打印此页面
打印此页面 收藏
收藏